Introduction
Can we determine the identity of a protein solely by resolving what binds to it?
In the DNA world, there is an analogous idea called sequencing by hybridization, in which the sequence of a DNA strand is mapped based on the identity of other DNA strands that bind to it. Although sequencing by hybridization did not gain much traction initially, many fluorescence, in-situ sequencing methods rely on hybridization and are extremely powerful. And our analogy runs further still.
One can run a protein-peptide binding assay on two proteins individually, then run it on two proteins in solution together. The difference in the “fingerprinting” signal for the combined proteins describes the occluded surface where the two proteins interact. For DNA, a method called subtractive hybridization can identify a DNA sequence that is missing between a control and test sample by making their cDNAs bind to each other — much like a diff command for computer files.
Proteins comprise over half of non-water weight in cells and their job is to interact with other biomolecules in a crowded, cellular environment. Solution phase protein-protein interactions are still poorly understood, especially the 15-40% that involve a globular protein recognizing a short linear peptide motif of several amino acids (Blikstad et al. 2015)
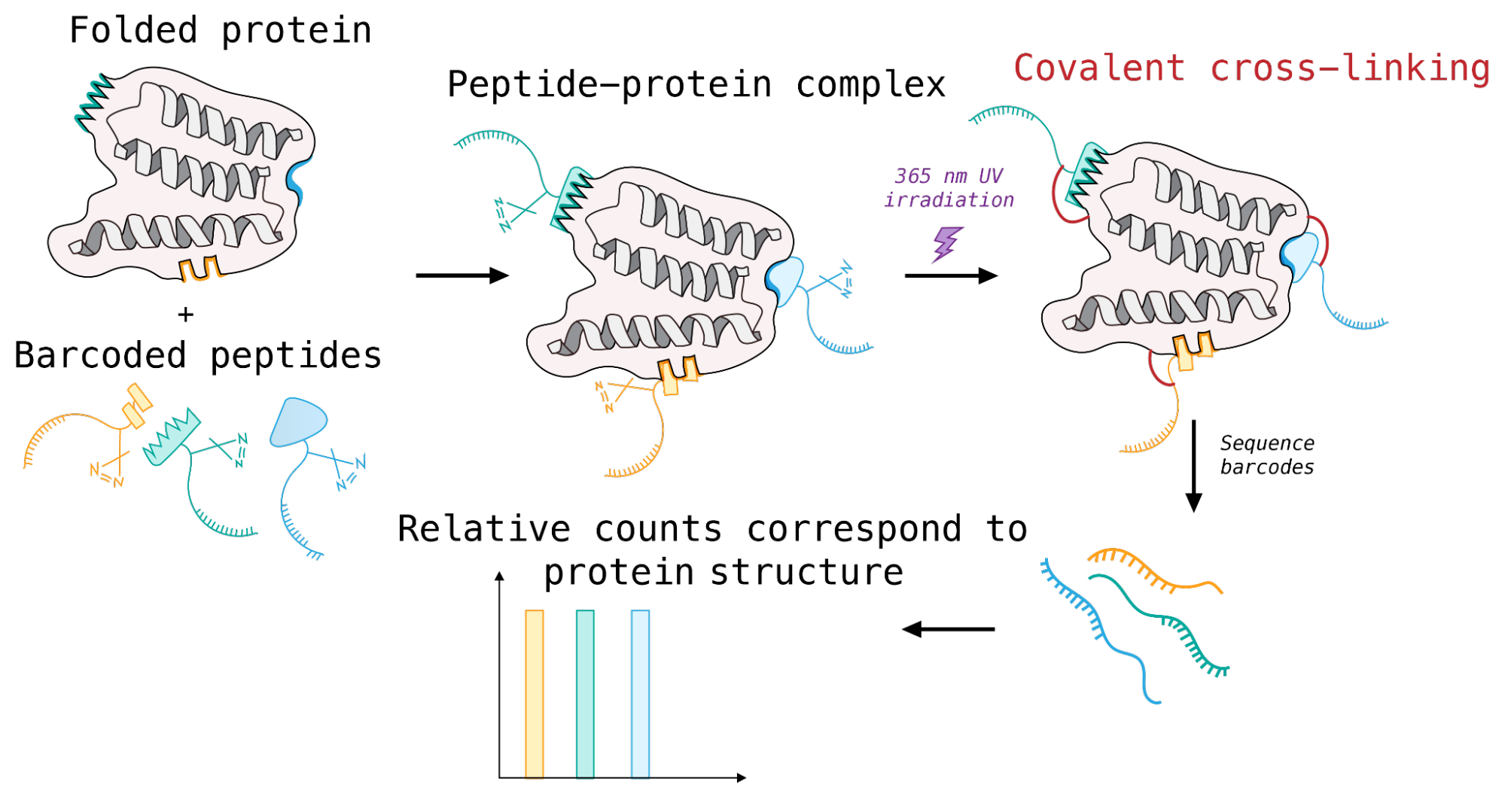
My aim this summer was to map the surface of individual proteins — their holes, pockets, and clefts — by quantifying each of their binding affinities to a large reference pool of small peptides fused to RNA barcodes. By sequencing the RNA barcodes of the peptides that stick to a protein, and mapping those to a peptide sequence, I planned to build a ‘fingerprint’ for various proteins. Such a platform would generate datasets that could help refine predictive algorithms for designing proteins, and could supplement existing methods for discovering weak protein binders that could be crucial in signaling pathways without knowing these interactions a priori.
Here I discuss the idea of fingerprinting proteins by binding affinities and its curious parallels to DNA sequencing methods. I will then introduce the progress I made this summer towards developing this tool.
Project Overview
Project Modules
Module 1: Design and geometry
Peptide-protein interactions are more difficult to predict than DNA-DNA interactions. There is no diff function for protein structures (yet). Simulations of DNA binding and secondary structure are nearly perfect, but predicting protein binding is daunting. DNA is composed of just four nucleotides. Proteins are complex, three-dimensional shapes composed of 20 amino acids.
Despite their complexity, protein binding affinity fingerprinting has been done, albeit with a small panel of 122 small molecule inhibitors (Kauvar et al 1995)
mRNA display, developed in 2001, is a clever method that uses in vitro transcription and translation to conjugate each displayed protein to its own corresponding mRNA. mRNA display libraries have also been used to create maps of protein-protein interactions (Johnson et al 2020)
Many display methods traditionally focus on finding high-affinity binders, while I wanted to map the distribution of weak interactions and fingerprint proteins based on these data.
I sought to combine some of these methods, together, by incorporating a cross-linker in the peptide mRNA library. The resulting molecules would be trifunctional: peptide, cross-linking moiety, and RNA barcode. After incubating a peptide library with a given protein sample, UV irradiation would crosslink the peptides to their target protein and form a covalent bond. Even transient interactions are preserved. Unbound peptides are washed away. The RNA barcodes covalently bound to the protein sample are then reverse transcribed and sequenced; the relative fraction of each barcode corresponds to the binding affinity between each peptide in the mixture and the sample protein.
Peptide-oligo conjugation
I began by conjugating peptides to single-stranded DNA, rather than doing mRNA display. This would serve as a starting point to test binding assays, explore the effects of a nucleotide tag on peptide-protein binding, and would also allow me to experiment with cross-linking without having to create an mRNA display library first.
I chose to use Growth factor receptor-bound protein 2 (GRB2) protein as my proof-of-concept protein to fingerprint. This protein is known for its role in signal transduction. It contains an SH2 domain and two SH3 domains (Src Homology 2/3) which are both common, conserved domains of about 100 amino acids that recognize other proteins that have a linear region containing a phosphorylated tyrosine. The stoichiometry of one SH2 domain to every two SH3 domains and its ability to recognize specific phosphorylated peptides makes it an appealing target for testing my fingerprinting method.
I constructed peptide-oligo conjugates to test if a known binding peptide motif — without its scaffold and with a comparatively large nucleotide tag — could bind to the SH2 domain in GRB2. Thus, I conjugated single-stranded DNA to a peptide with a phosphorylated tyrosine (DDPSpYVNVQNLK), synthesized by Genscript, as well as a similar DNA strand to a version with a non-phosphorylated tyrosine (DDPSpYVNVQNLK). The peptides were modified with a C-terminal lysine to introduce a conjugation site. Both DNA oligos were also labeled with Cy5, a fluorophore, on the 3’ end and synthesized by Integrated DNA Technologies. One oligo mimicked the DNA coding for the peptide, and the other was the same but with four point mutations that prevent off-target binding to the two strands’ complement sequences. Sequences are listed in the Appendix ).
I conjugated the oligos and peptides using copper-free click chemistry — a simple reaction that proceeds quickly across a wide range of solvent conditions — and followed a protocol that was originally designed to conjugate oligos to antibodies. The click chemistry involved methyltetrazine (mTz) and trans–cyclooctene (TCO) click chemistry; briefly, both groups were attached to a spacer arm and N-Hydroxysuccinimide (NHS) ester. NHS esters target primary amines — a single carbon bound to an NH2 group. I labeled the 5’ amine modified DNA oligo with TCO-PEG4-NHS by reacting it in a pH 8.4 borate buffer and 10% DMSO in two sequential additions. The reactions were quenched with addition of 1M glycine, and desalted with Bio-Rad Micro Bio-Spin P-6 columns.
To verify that the conjugation and labeling were successful, I used a BioAnalyzer RNA pico chip, a chip-based gel electrophoresis platform. The kit dye binds to single stranded DNA as well as RNA, and thus I could observe a distinct size shift after the modification and observed a high conjugation efficiency of >80%.

I labeled the peptides with mTz-PEG4-NHS. The NHS targets either the C-terminal lysine or the N-terminus of the molecule. Buffer exchange into the borate buffer was performed with Microsep Advance Centrifugal Devices with Omega Membrane 1K (PALL corporation) for 90 minutes. The labeling reactions were performed again in pH 8.4 borate buffer containing 10% DMSO and quenched with 1M glycine. Another 90 minute spin was performed to purify the sample.
The same RNA pico assay allowed me to confirm that the peptide was successfully conjugated to the DNA as well, albeit with a lower efficiency. At the end of the summer, I had produced the conjugates but was still troubleshooting the pull-down binding assay with His-tagged (a polyhistidine tag that binds to nickel and cobalt and commonly used for purification) GRB2 and the Cy5-labeled peptide oligo conjugate molecules.
Module 3: mRNA display and library design
Since individually conjugating DNA to peptide is laborious and low-throughput, I aimed to use mRNA display to synthesize in vitro a library of peptides, each one conjugated to its own RNA. Thus, it would be possible to reverse transcribe the RNA and then perform next-generation sequencing to quantify those peptides that bound to a target protein.
mRNA display begins a DNA sequence of the protein or peptide desired, surrounded by a T7 transcription promotor, a ribosome binding site and start codon, and a 3’ constant region that enables ligation to a linker downstream. The DNA can be obtained from a cell’s mRNA / cDNA for uneven distribution libraries, or it can be chemically synthesized. Then, one performs in-vitro transcription, enzymatically digests the DNA, and purifies the RNA.
The RNA is next ligated to a short DNA linker with a 3’ puromycin. Various linker designs are available — a GC-rich hairpin-forming linker with biotin incorporated as well as inosine for cleavage (Johnson et al. 2020), as well as a simpler poly(A) linker that can be targeted with oligo (dT) beads (Barendt et al. 2013). The 3’ end of the RNA is ligated to the 5’ phosphorylated DNA linker.
During in vitro translation of the modified RNA, each strand of the RNA will have a short single-stranded DNA linker on its 3’ end. Translation proceeds from 5’ to 3’ until the ribosome hits the linker, then the puromycin eventually diffuses into the A site of the ribosome. The puromycin feigns as a tRNA, and it is accepted and incorporated into the nascent polypeptide chain. This releases the ribosome from the strand, and the 3’ end of the single-stranded mRNA-DNA conjugate is thus covalently bound to the C terminus — the ‘end’ of the protein or peptide chain.
I have successfully performed in vitro transcription with several DNA template strands encoding FLAG and HIS tags. After optimizing the DNA-RNA ligation and translation process, I am planning to add a consistent, photoactivatable crosslinker to each molecule. These crosslinkers will be added to the ssDNA linker. I have ordered DNA linkers with internally modified amines at various positions for this application, allowing for a one-to-one correspondence between peptide and crosslinker. Possible choices include NHS-diazirine, which nonspecifically targets the peptide backbone, as well as BS3 or NHS-PEG4-NHS, which both target primary amines or lysines. This one-to-one correspondence between peptide and crosslinker will allow for a reproducible cross-linking method.
To start out, I’ve designed and ordered the DNA for a pooled peptide library containing several thousand human proteome peptides. These peptides range in length from 8 to 34 amino acids. The 5’ end of each DNA sequence contains a constant region with a T7 promoter, E. coli ribosome binding site, and ends with a start codon. There is also a 3' constant sequence, and the 3’ end will be ligated to a ssDNA puromycin linker. The 3’ constant region contains a GGS linker, a PvuII cleavage site for optional exclusion of the His tag pre-transcription, a 6xHIS tag, and another GGS linker. Crucially, this version has no stop codon. I have also ordered DNA with similar 5’ and 3’ constant regions, but without the PvuII cleavage site and with randomized (NNB) amino acids.
Conclusion
Above all, this summer I learned how to set up and start a project. And to be brave. I dove deeply into the literature and found various mRNA display protocols as well as RNA-DNA ligation protocols. I found a cool paper describing a method to search for ice-binding peptides using phage display, which was inspired by deep-sea fish and cold-climate frogs that evolved their own antifreeze proteins to survive in harsh conditions. I also learned about computational efforts to predict peptide-protein binding that focus on conserved domains and weak interactions. Such computer algorithms are inherently limited by data; we simply don’t know enough about protein structures and their bindings to predict how things interact inside of cells. For a great discussion on this topic, see Derek Lowe’s recent blog post on the stickiness of the protein and the tube.
There are many open questions for my project, too: Is there a universal set of high-affinity peptide binders that will allow us to characterize an unknown protein or unknown sample? How can we span the enormity of protein space with binders such that we can recognize every protein? Can we identify proteins solely by measuring their weak or moderate interactions, or are strongly binding peptides also required?
My project started as an image of a protein decorated at its binding pockets by peptides — a peptide library of flexible LEGOs binding to a larger LEGO structure. I don’t know how to reason about forces and interactions at the smaller scales that are relevant to biology — physics doesn’t really teach you and neither does biology. What does environment or affinity or temperature even mean when the world gets that small? Protein fingerprinting is a satisfying compromise: it can be messy and there can be bias, but as long as your mess and bias are consistent, you will still hopefully learn something about your system.
I’ve gotten less interested in finding strong binders and more interested in weak and transient interactions. My dream would be to assemble a large library of barcoded peptides that can form a distinct binding signature for any human proteome protein we know of. t’d theoretically be able to detect some forms of structure-disrupting mutations, too. It could be used to fingerprint proteins, but also cells and cell surfaces. Furthermore, if you can link mRNA to protein in vivo (in-cell mRNA display, really) you could get a one-to-one mapping between signal and reporter tags in vivo.
While applications are cool, I’m most fascinated by basic questions; how matter interacts with other matter on the scale of a cell, and in deciphering the wigglings and jigglings of small molecules and proteins.
