Introduction
This summer, I explored whether Chlamydomonas reinhardtii, a species of green algae, could serve as a starting point for engineering a free-living chloroplast. In the process, I was searching for a new window into one of the most compelling mysteries in biology: the endosymbiotic events that turned free-living bacteria into captive organelles within a larger host about two billion years ago.
One such event produced mitochondria, provoking the evolution of eukaryotic life. Half a billion years later, a second event
We can make well-reasoned guesses, based on complementary observations and corroborated by mountains of phylogenetic data. On the other hand, generating testable hypotheses is a bit harder. Endosymbiosis reappears throughout nature, from corals to insect guts, but it almost never leads to the generation of new organelles like chloroplasts. That is only thought to have occurred a handful of times in the history of Earth. As a result, no one can claim that our understanding of these important, endosymbiotic events are anywhere near complete. Ask anyone to recreate a stable endosymbiotic event in a test tube, and their results will pale in comparison to nature’s handiwork (although recent progress on this front has been exciting).
As I argued earlier, experimental advances have historically been impeded by the limitations of available tools and technology. This hasn’t been for lack of effort. In the 1920s, American biologist Ivan Wallin sought to prove that mitochondria were descended from free-living bacteria by isolating them in culture. He claimed success, but his results proved irreproducible. Of course, this could never work — modern mitochondria have been coevolving within their host cells for so long that the two are completely inseparable. More precisely, it could never work without first modifying the mitochondria to prepare them for life outside their host. In Wallin’s time, years before the first experiments linking DNA and heredity, this was impossible. Today, however, the tools of synthetic biology give us the power to design, edit, and rewrite genomes. In other words, perhaps it’s time to try again.
{kind=link}
Project Overview
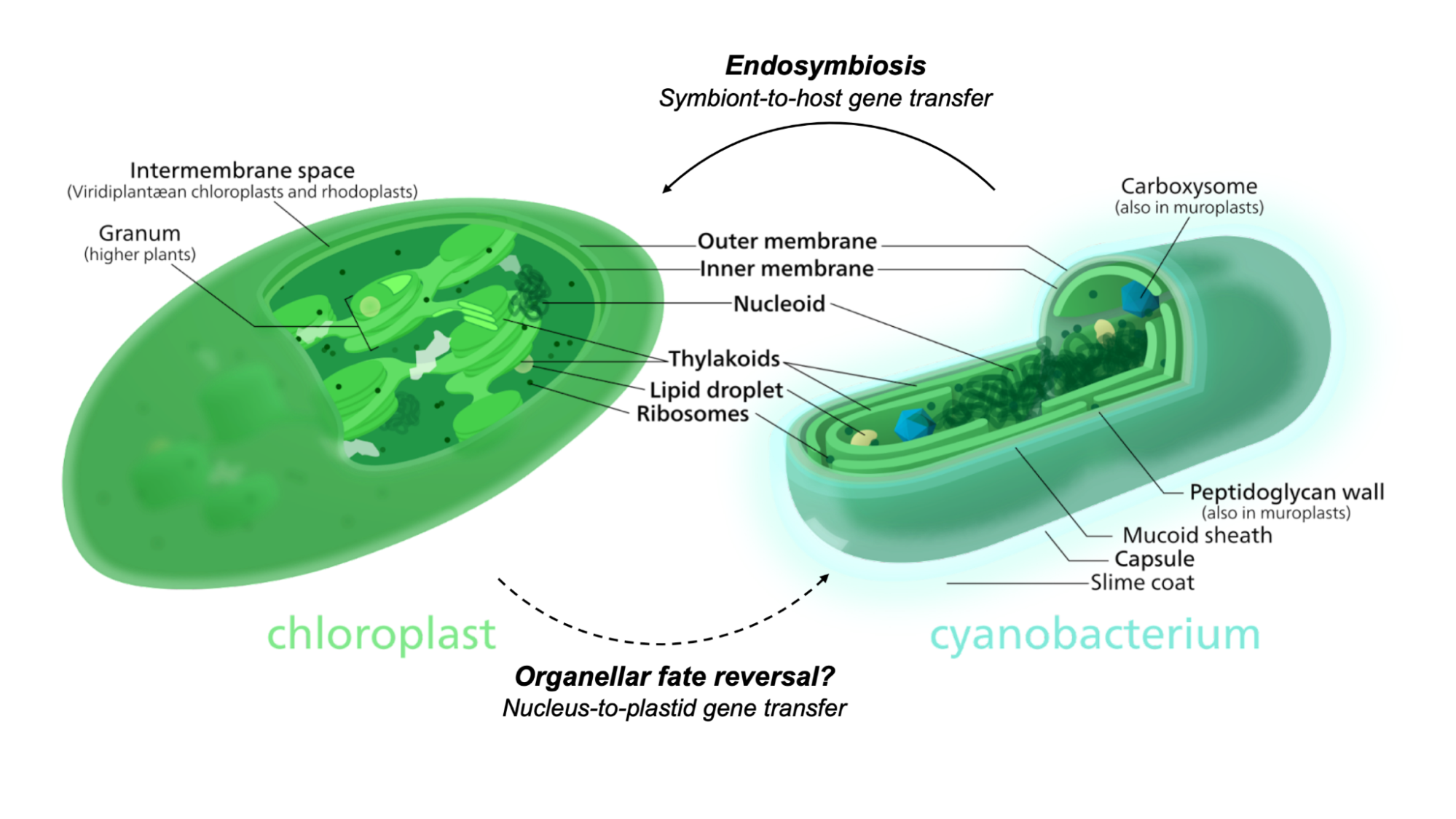
The evolutionary transition from a free-living cyanobacterium to a “domesticated” chloroplast is marked by numerous gene transfer events. Over countless generations, the endosymbiont gradually lost functions that were no longer needed within its newfound intracellular niche. Some genes moved into the host’s genome, placing it under host regulatory control. I reasoned that repatriating these genes would restore lost functions to the organelle — and eventually rewind the evolutionary clock to create a free-living chloroplast similar to its ancestral cyanobacterium (Figure 1). My initial goal was to evaluate the promise of this approach by identifying candidate genes, transforming them back into the chloroplast, and assessing the results. While I encountered several technical and conceptual bottlenecks, I still learned useful things along the way: I tested some current tools and techniques for organelle manipulation, designed a new protocol for working with isolated chloroplasts ex vivo, and left the laboratory brimming with ideas for future work.
My project's milestones are broken into 4 discrete modules:
- 1. Designs and planning,
- 2. Chloroplast imaging,
- 3. Chloroplast transformations, and
- 4. Chloroplast life support.
Project Modules
Module 1: Design and planning
My first few weeks in Boston were largely spent preparing the reagents and equipment I’d need for my experiments. I began with three considerations: selecting an appropriate host strain, establishing an efficient culture system, and devising the tools I’d need to study chloroplasts and their viability ex vivo.
A wide variety of growth media compositions are available for culturing algae and cyanobacteria. In Chlamydomonas research, one common general-purpose recipe is tris-acetate-phosphate (TAP); a variation with no carbon source, TP, is used for phototropic selection. Traditionally, TAP is supplemented with Hutner’s trace elements, but preparing this mixture in-house takes 1-2 weeks and is highly sensitive to small changes in pH.
I chose CC-400, a classic cw15 (cell wall-deficient) mutant, as my workhorse strain of C. reinhardtii. The chloroplast isolation and transformation protocols I would be using called for cell wall removal by gametolysin treatment, but working in a cell wall-deficient strain would allow me to skip that step and save time.
To provide steady, uniform illumination under controlled conditions, photosynthetic organisms are typically cultured inside of specially-designed lighted incubators. In the case of microalgae like C. reinhardtii, which can be grown in suspension, a photobioreactor may be used. I didn’t have convenient access to this equipment, but there was a DIY solution sitting disused in a corner of the lab. This homemade incubator rack — a shelf holding an orbital shaker, lit by programmable fluorescent lamps — was constructed by a former postdoc and became my base of operations. I added extra shelving to accommodate my flasks. Soon, I had all the basics needed to cultivate my cells (Figure 2). It didn’t have features like CO2 fixme or temperature control, but it would get the job done. For my dark mutant cultures, I repurposed an old benchtop shaker by wrapping the lid in foil (not pictured).

Module 2: Chloroplast imaging
Before diving into experiments, I wanted to find my footing — after all, I’d never worked with microalgae before. I wanted to build what Barbara McClintock called "a feeling for the organism”: a sense of intuition for how C. reinhardtii grows and behaves. For me, that required getting eyes on cells up close.
Once I had liquid cultures growing, I viewed them under a 40X optical microscope — just close enough to resolve individual cells as tiny green circles, but insufficient to make out any details. While the CC-400s appeared static, the CC-5168s were wonderfully motile; since they’d been growing in the dark, illuminating them under the objective triggered a flurry of phototaxis. To measure cell growth, I compared manual counts from a haemocytometer with readings from an automated cell counter, and found excellent agreement.
But I wanted to get a closer look. What kind of tooling exists for capturing detailed images of C. reinhardtii cells? I found beautiful fluorescent microscopy work online, but little of it seemed to focus on chloroplasts. In turn, this made me wonder about the state of the palette of chloroplast-compatible fluorophores available to algae researchers. After all, effective chloroplast engineering in Chlamydomonas will require reporters — for instance, to help assess the localization of a recently repatriated chloroplast protein!
My initial search of the literature seemed promising. In 2002, Franklin et al. reported the development of a green fluorescent protein (GFP) codon-optimized for expression in C. reinhardtii chloroplasts, which boosted protein accumulation by 80-fold. However, upon closer inspection, the authors acknowledge an important caveat: despite their improvements, GFP fluorescence “was not easily visualized in the chloroplast”. Instead, expression was only detectable by Western blots and gel assays!
The reason? The chlorophyll and carotenoid content of C. reinhardtii chloroplasts causes them to be highly autofluorescent in the same range of wavelengths that excite GFP, making green fluorophores generally ill-suited for imaging in the organelle. Instead, fluorescent tagging of Chlamydomonas chloroplasts has only been successful in other regions of the spectrum, such as with the yellow protein Venus. To investigate this myself, I reached out to the Nikon Imaging Core at HMS, who helped me view my algal cultures using confocal microscopy. We tested a wide range of wavelengths to survey the range of C. reinhardtii chloroplast autofluorescence (Figure 3A).
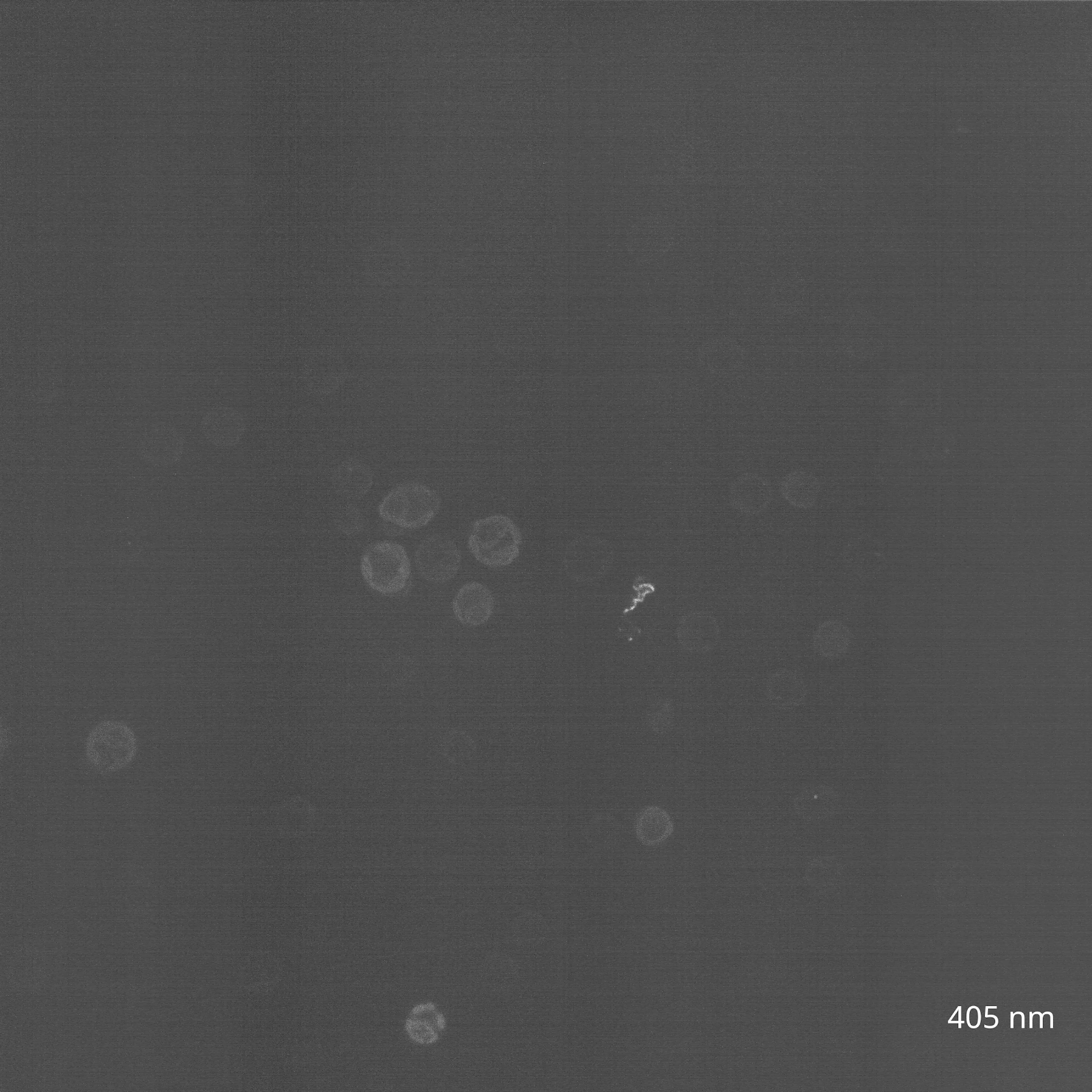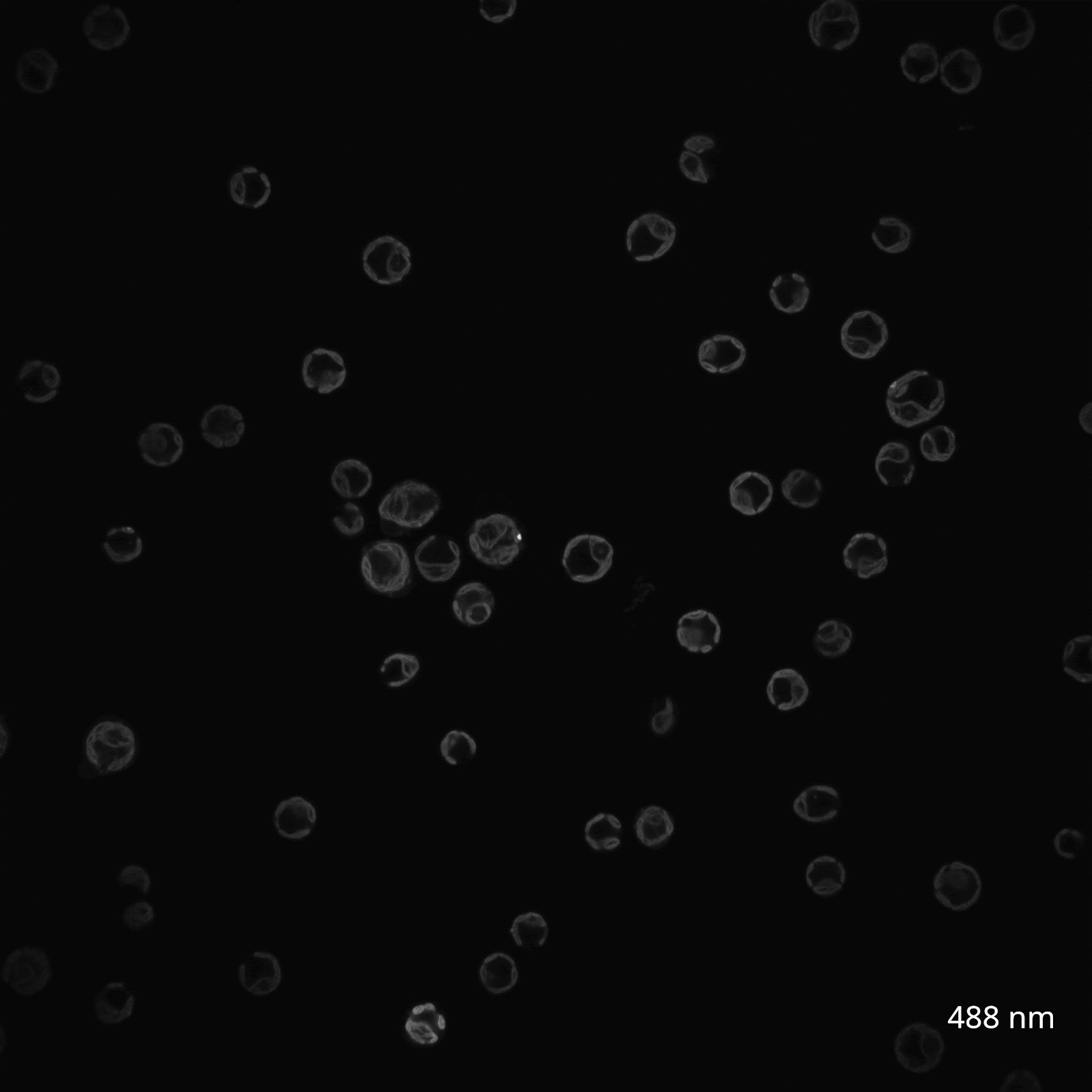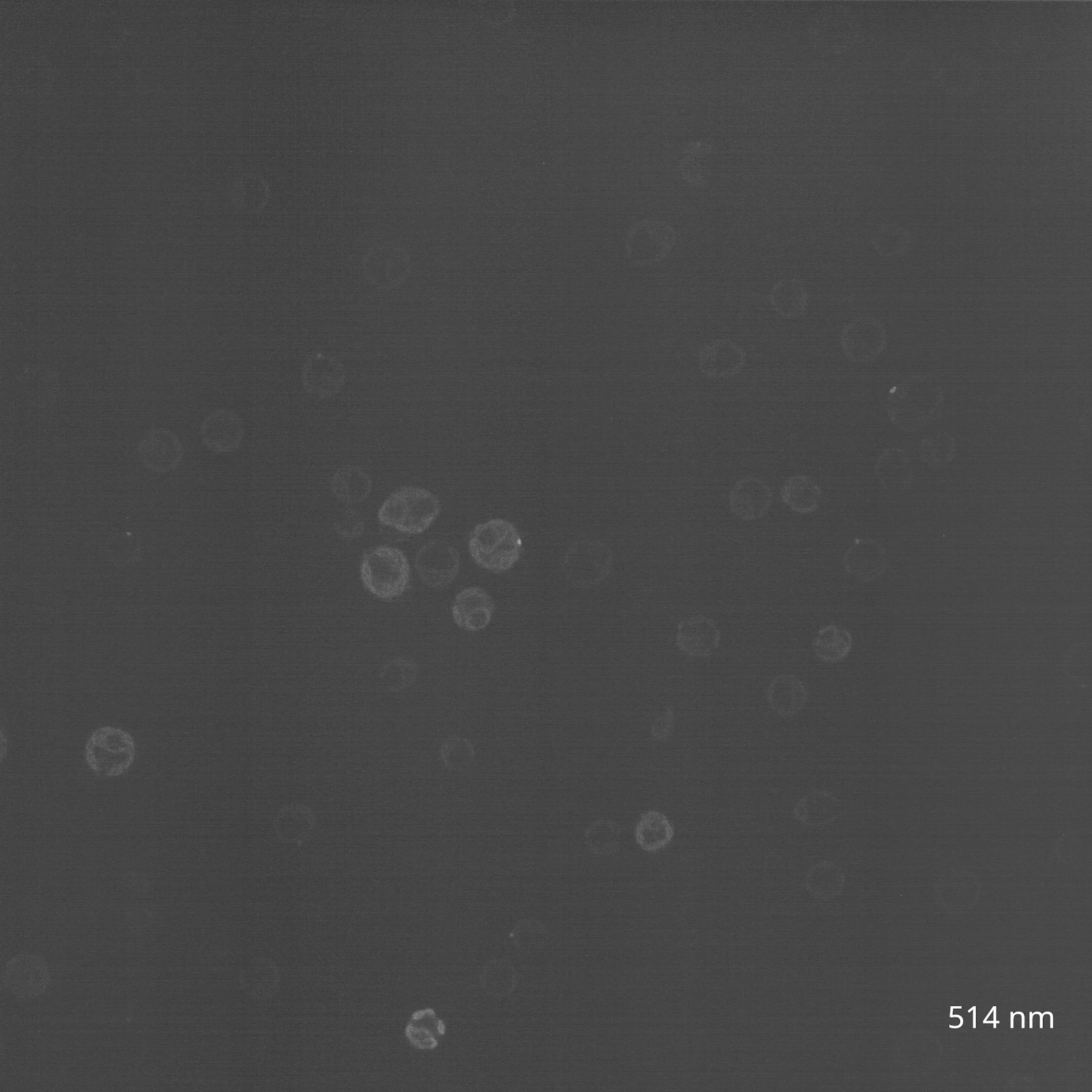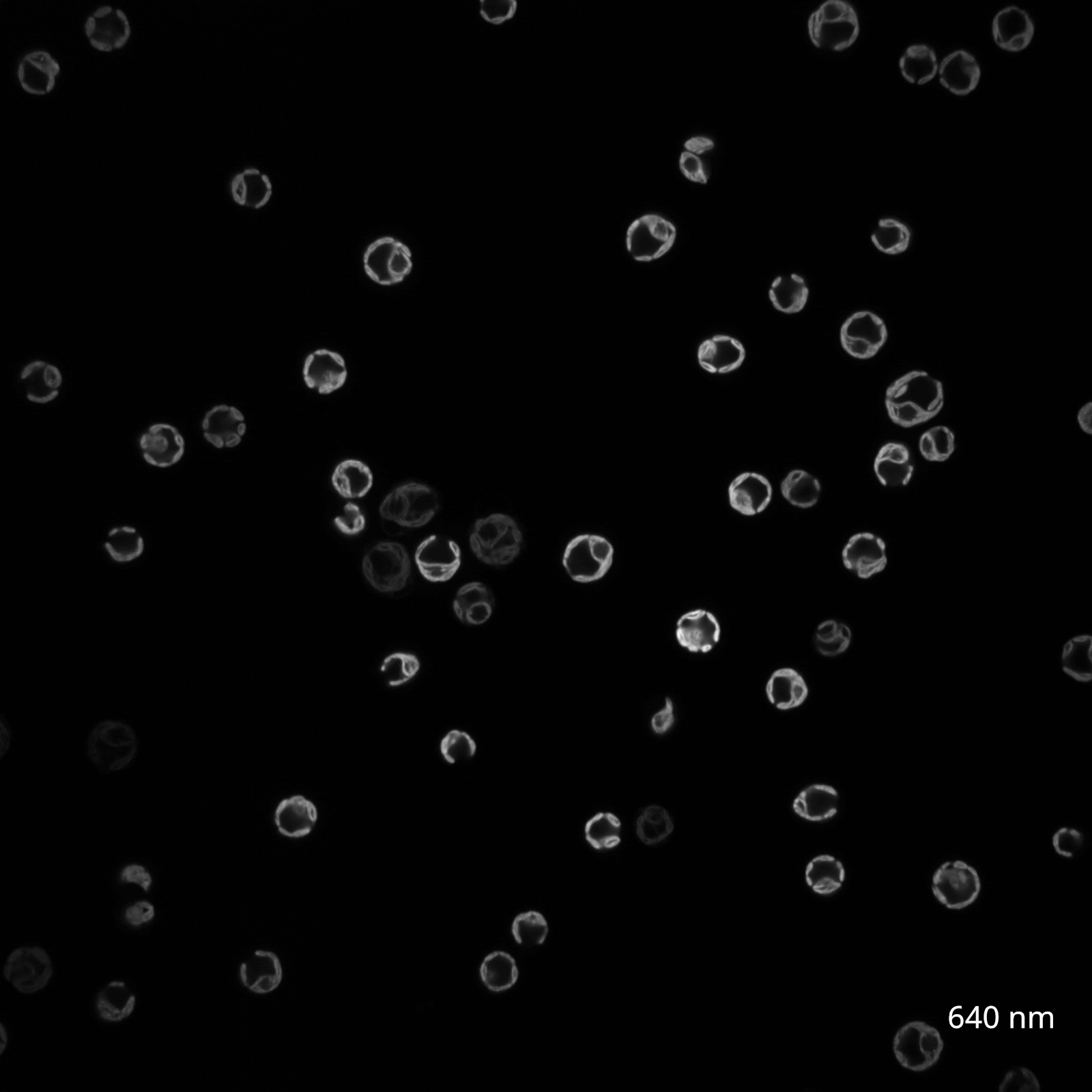
(B) CC-400 cells viewed on a spinning disk confocal in the far red channel (640 nm, 30s timelapse).
In contrast, yellow and red wavelengths seemed to offer a more promising signal-to-noise profile. After consulting with Talley Lambert, the creator of FPbase, I selected mCherry, mScarlet-I, and moxVenus as my palette of fluorophores to eventually test in C. reinhardtii chloroplasts.
Module 3: Chloroplast transformations
There are two principal bottlenecks to creating a free-living chloroplast by genetic engineering. The first is a question of information: what are the genes that need to be returned to the chloroplast in order to restore its autonomy? Some of them are now encoded in the nucleus, but many others have been lost entirely. We need a blueprint — whether that’s informed by extant cyanobacterial genomes, phylogenetic reconstructions, or some secret third thing. It’s a problem begging for computational solutions, but over the summer, I wanted to focus on maximizing my time at the bench. This leads to the second hurdle.
Even once we know exactly what to synthesize, chloroplasts are far less genetically tractable
Techniques that can adequately solve the delivery problem only emerged in the late 1980s with the development of biolistics, or “gene guns”, which enabled the first-ever transformations of yeast mitochondria and C. reinhardtii chloroplasts. However, there are also alternative methods that do not require expensive specialty equipment. One such technique involves a little vortexing; after removing its cell walls, a suspension of Chlamydomonas is mixed with donor DNA and small (~500 um) glass beads, then agitated briefly. This ostensibly introduces transient perforations to the membrane, allowing DNA to enter cells. This is effective for nuclear transformations, but it also seems to work on Chlamydomonas chloroplasts.
I transformed my CC-5168 cells following Economou et al.’s more recent take on the protocol. Recall that this strain is non-photosynthetic; more specifically, it carries a mutation inactivating the chloroplast psbH gene. The companion pSRSapI plasmid carries an intact psbH gene, along with a cloning site for inserting a gene of interest. Since chloroplast DNA repair proceeds by homologous recombination, successful transformation will rescue the cells’ photosynthesis, allowing them to grow without acetate supplementation. As an initial positive control, I ran my transformation with the empty pSRSapI vector. (I also attempted to insert codon-optimized versions of mCherry, mScarlet-I, and moxVenus in parallel, but my cloning failed repeatedly and I was unable to diagnose the cause, so I put that plan on hold).
And then I waited. The protocol claimed that recovering transformants could take 3-4 weeks, which was already quite slow. It ended up taking twice as long — almost 8 full weeks — before the first green colonies of C. reinhardtii appeared, by which point I had already left Boston. The lengthy iteration time indicated that focusing on chloroplast transformations might not be the best use of my time.
While brainstorming ways to improve transformation efficiency, I stumbled across a paper by Mason et al. describing a method for isolating intact chloroplasts from C. reinhardtii cells. At first, I wondered if attempting to transform chloroplasts ex vivo might be more effective. Rather than shuffling DNA through two sets of membranes (first the eukaryotic host’s, then the chloroplast envelope), attacking the organelle directly might be easier. Contending with only one kind of structural composition, rather than two different sets, could simplify the needed chemistry. In turn, however, this raised many more new questions: once I’ve transformed a chloroplast outside a Chlamydomonas cell, how do I get it back in? And in the meantime, could I even keep the isolated organelle “alive”?
This led me to a new question: how much progress could I make towards growing free-living chloroplasts without doing any genetic engineering?
Module 4: Chloroplast life support
Once extracted from its host cell, an intact chloroplast should survive
Using equipment I already had on hand, I started devising a microplate-based assay using the concept of chloroplast autofluorescence. When a chlorophyll molecule in the antenna complex captures energy from incident light, it is transferred to the photosystem to drive photosynthesis, with the remainder dissipated as heat and light. Thus measuring chlorophyll fluorescence can provide a readout of photosynthetic activity, which is the principle underlying techniques such as pulse-amplitude modulation (PAM) fluorometry. Moreover, the chlorophyll biosynthesis genes in C. reinhardtii are (today) nuclear-encoded, meaning that chlorophyll production should cease at some point after chloroplasts are separated from their hosts. Hence, comparing time-series measurements of chlorophyll autofluorescence between whole cells and isolated chloroplasts might expose differences in organelle lifetime. Beyond simply assessing the baseline survival of isolated chloroplasts, I also aimed to extend it by tweaking their environment in vitro.
I was inspired by the reconstitution experiments of classical developmental biology, in which cellular components are combined piecewise in order to recapitulate biological phenomena. If extracting a chloroplast will separate it from essential host factors, then could bathing it in a cocktail of those factors help it survive? I reasoned that treating isolated chloroplasts with bulk cytoplasmic extract might act as a kind of organelle life support system, and I hoped the difference would be visible using my assay.
Following Mason et al.’s method, I extracted C. reinhardtii chloroplasts by forcing whole cells through a narrow-gauge syringe needle. I performed the extraction in the evening, around the day-night transition in my incubator. After several rounds of centrifugation through sucrose gradients (Figure 4A), I purified the resulting lysate to obtain a suspension of (hopefully) intact chloroplasts, as well as a cytoplasmic fraction. I loaded everything into a 96-well plate: whole cells in TP, isolated chloroplasts, and isolated chloroplasts supplemented with cytoplasmic extract, plus all the necessary controls. I then tracked their chlorophyll fluorescence every 2 minutes for the next 10 hours (Figure 4B). For a detailed protocol, see the Appendix.
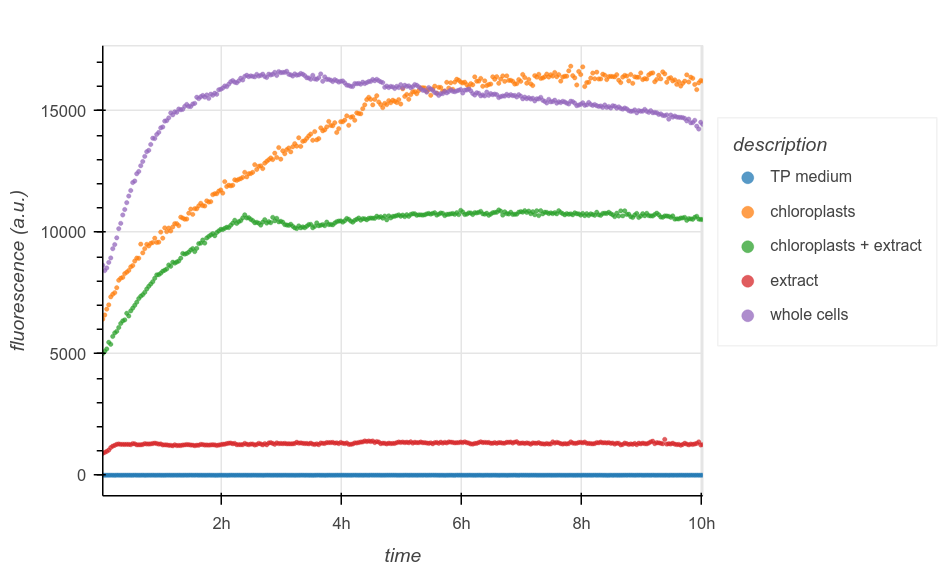
The results were exciting but inconclusive. As expected, the negative controls (TP or cytoplasmic extract alone) exhibit low, constant fluorescence. Whole cells increase in signal sharply over the first few hours, then plateau; if fluorescent intensity is correlated with chlorophyll biomass, then this could be consistent with the flurry of mitotic divisions that occurs during the day-night transition in synchronous culture, before entering G0 for the rest of the dark phase. Confusingly, though, both chloroplast conditions also experience an increase in fluorescence. This could suggest that they are contaminated with unbroken cells; it may also indicate other confounding factors in my fluorescence measurements.
Note that it’s difficult to compare the positive control to the chloroplast wells directly, since while I used a known quantity of cells as input in all cases, I did not quantify the yield of isolated chloroplasts.
One idea for future work is to combine this method with complementary measurements, like using a Hill reagent to track the redox activity of Photosystem II. Because this provides a spectrophotometric readout of photosynthesis, it can be added directly to my assay by simply programming an extra step into the plate reader. This would help reveal whether my fluorescence measurements are representative of chlorophyll biomass, photosynthetic activity, or something else entirely. Combined with a series of other improvements to make my assay more quantitatively powerful (some of which are noted below), this approach may ultimately help answer whether and how chloroplasts can survive outside of their host cells.
Conclusion
So, whither the chloroplast? This summer, I set out to engineer a free-living organelle by re-endowing it with lost genes from the ancestral endosymbiont. And while I didn’t get there, my preliminary investigations have yielded some exciting new ideas.
I surveyed the current state of knowledge in the field, and identified what I believe are important bottlenecks and directions for future work. For example, I tested some existing technologies for manipulating chloroplasts, and encountered some of their shortcomings firsthand. This shifted my immediate interests away from specific genetic manipulation, and towards more systems-level approaches.
To this end, I began work on a simple and scalable assay which (with continued development) will allow researchers to interrogate organelle biology in new ways. To my knowledge, the key idea — using reconstitution-style experiments to test the minimal set of conditions required for organelle autonomy — hasn’t been seriously explored since Ivan Wallin’s work a century ago.
Overall, while I have a high-level sketch for a chloroplast life support assay, more work is needed to make the assay useful and quantitative. The isolation and measurements need to be carefully repeated at well-defined stages in the C. reinhardtii cell cycle to help make sense of the resulting data. Concentrations in every well should be measured first, perhaps with an initial spectrophotometric step in the plate reader, rather than simply using equal volumes of input. The isolation protocol needs to be repeated and validated to independently assess chloroplast yields, purity, and quality. It is also likely that my measurement scheme (e.g. the intensity of incident light) requires fine-tuning, in case it leads to confounding results such as triggering a photoprotective response.
Looking ahead, there is still so much to do. I’m left asking even more questions than I started with. Besides iterating on the ideas mentioned here, I still intend to test computational approaches which should yield complementary results — for example, by attempting to retrace the evolutionary history of chloroplast gene transfer events, in order to guide future engineering work.